
De Nobelprijs voor Scheikunde 2013 is toegekend aan Martin Karplus, Michael Levitt en Arieh Warshel. Zij zetten belangrijke stappen in de ontwikkeling van methoden om chemische reacties te kunnen modelleren met behulp van de computer. Het drietal ‘bracht het experiment naar cyberspace’, aldus het Nobelprijscomité. Dat scheelt niet alleen veel labwerk, maar leidt ook tot nieuwe geneesmiddelen, betere zonnecellen en efficiënte chemische processen.

Chemici gebruiken de computer misschien wel vaker dan hun labjas.
Purdue UniversityToegegeven, het beeld van de chemicus in zijn (of haar) witte jas klopt nog steeds. Alleen in het laboratorium kun je écht experimenteren. Maar scheikundig onderzoekers brengen vandaag de dag óók heel veel tijd door achter de computer. De theorie helpt de praktijk – en andersom.
Al zolang scheikundigen met hun reageerbuizen en reactiekolven in de weer zijn, proberen ze te begrijpen wat er precies gebeurt. En omdat je moleculen niet kunt zien, maken ze gebruik van modellen. Stokjes en bolletjes helpen zicht te krijgen op de structuur en de reactiemogelijkheden: welke moleculen of molecuulgroepen zouden met elkaar kunnen reageren? En hoe gaat dat dan? Niet voor niets was het eerste model van DNA een enorme doorbraak in het begrijpen van de werking van het complexe molecuul.
Chemie in de computer
Natuurlijk is zo’n stokje-bolletje model een grove versimpeling van de werkelijkheid. Echte moleculen bewegen razendsnel, vibreren, draaien. En ze reageren in een flits, die voorbij is voor je er erg in hebt. Krijg daar maar eens grip op.
Gelukkig is er tegenwoordig de computer. Computational chemici kunnen allerlei mogelijke reactiemanieren simuleren, het meest voor de hand liggende reactiemechanisme onderscheiden en de rol van allerlei atomen tijdens de reactie vaststellen.
De winnaars van de Nobelprijs voor de scheikunde van dit jaar hebben hier een cruciale bijdrage aan geleverd. Zij hebben er voor gezorgd dat computers, die immers niets méér zijn dan hele snelle rekenmachines, met chemische processen kunnen omgaan.
Kwantummechanica
In feite zijn alle computermodellen wiskundige constructies waarin alles uit de ‘echte’ wereld is teruggebracht tot formules. In de chemie is het niet anders. Hoe atomen en moleculen met elkaar in interactie gaan, of ze elkaar aantrekken of afstoten, hoe ze door water heen bewegen, het is allemaal vast te leggen in formules. Alleen: als het om de chemische details gaat – en vooral als er reacties in het spel zijn – dan kan dat tamelijk ingewikkeld worden. Dan moeten de computerchemici namelijk een beroep doen op de kwantummechanica en kunnen ze niet meer volstaan met de ‘klassieke’ beschouwing van de chemische verschijnselen.
Dit is ook bekend uit het heelal: iets elementairs als de beweging van planeten in een zonnestelsel is goed te beschrijven op basis van de klassieke natuurkunde van Kepler en Newton. Maar voor de finesses zijn de inzichten van Einstein en zijn tijdgenoten onontbeerlijk.
Rusttoestand
De klassieke modellen geven prima zicht op bijvoorbeeld de structuur van eiwitten. Onderzoekers zien welke delen star zijn, welke flexibel en hoe de eiwitten gevouwen zijn. Een groot nadeel van deze aanpak is echter dat het molecuul in een soort ‘rusttoestand ‘ wordt gemodelleerd. In kwantummechanische termen zegt men: in de toestand met de laagste energie. De klassieke modellen gaan voorbij aan het feit dat elektronen in een molecuul een heel eigen leven kunnen leiden en in allerlei energietoestanden kunnen voorkomen.
Vooral wie meer wil weten over de activiteit van moleculen kan niet om de kwantummechanica heen. Maar zou je louter op basis van díe theorie willen voorspellen hoe een molecuul reageert, dan ben je wel even bezig. Je moet alle elektronen modelleren, alle atoomkernen waar ze omheen cirkelen en alle mogelijk configuraties van een molecuul. Zelfs voor relatief eenvoudige moleculen is dat onbegonnen werk. En dan hebben we het nog niet eens over de moleculen die ons leven beheersen – letterlijk – zoals eiwitten en enzymen. Die bestaan soms uit wel honderdduizenden atomen. Daar kom je ook met de krachtigste supercomputers niet uit.
De winnaars van de Nobelprijs van dit jaar worden vooral gelauwerd vanwege de manier waarop ze de brug hebben geslagen tussen modellering gebaseerd op de klassieke Newtoniaanse benadering en de modellering volgens de principes van de kwantummechanica. Of, in de woorden van het Nobelprijscomité: Dankzij het werk van de laureaten zijn in de computational chemistry zowel de appel van Newton als de kat van Schrödinger terug te vinden.

Retinal.
Wikimedia CommonsRetinal
Het eerste belangrijke wapenfeit van de Nobellaureaten was de modellering van het eiwit retinal door Arieh Warhel en Martin Karplus. Dat die hun vizier op dit relatief eenvoudige eiwit richtten was niet toevallig. Het molecuul heeft een duidelijk waarneembare relatie tussen kwantummechanische en klassieke eigenschappen: onder invloed van licht komen elektronen in retinal in een energierijke, aangeslagen toestand terwijl tegelijkertijd de structuur van het molecuul verandert. Precies daardoor vervult retinal een belangrijke functie in het menselijke netvlies (de retina): het ligt aan de basis van ons ‘zien’.
In het onderzoek naar retinal vulden Warhel en Karplus elkaar perfect aan. De eerste had bij het Weizman onderzoeksinstituut in Israël samen met Michael Levitt één van de eerste computerprogramma geschreven om tamelijk ingewikkelde moleculen te modelleren. Het was gebaseerd op de klassieke aanpak. Karplus, van Harvard University, was juist gespecialiseerd in het modelleren van chemische reacties met de kwantummechanische methode.
Enzymreacties
Met hun gezamenlijke publicatie over retinal in 1972 legden de beide Nobellaureaten voor het eerst een chemisch relevante koppeling tussen kwantummechanisch en klassiek fysisch modelleren. Vooral Warhel had daarna de smaak te pakkken. Hij zocht contact met zijn oude studiegenoot Levitt, die inmiddels aan het werk was bij Cambridge University (Verenigd Koninkrijk), destijds wereldleider in het modelleren van biologische moleculen. Samen stelden ze zich een ambitieus doel: het modelleren van enzymen – de eiwitten die verantwoordelijk zijn voor vrijwel alle reacties in levende organismen.
Het kostte Levitt en Warshel vervolgens vier jaar om de aansluiting tussen beide modelleringsmethodieken te verfijnen. In 1976 publiceerden ze het allereerste computermodel van een enzymatische reactie. En dat niet alleen: hun model was voor elk willekeurig molecuul geschikt te maken. Groot of klein, ingewikkeld of eenvoudig, dat maakt eigenlijk niet uit. Het is een revolutionaire aanpak gebleken, die tot op de dag van vandaag zijn vruchten aflevert.
Het beste van twee werelden

De strategische aanpak van computational chemici is om computerkracht dáár in te zetten waar het ‘t meest oplevert. Voor de elektronen en atoomkernen die direct een rol spelen in de chemische processen voeren ze gedetailleerde kwantumfysische berekeningen uit. Minder ’actieve’ delen van het molecuul krijgen een klassieke behandeling. En voor de afgelegen, minst interessante gebieden van het molecuul is het zelfs mogelijk om alle atomen op één hoop te gooien en die als één ‘klomp’ materie te beschouwen, als diëlektrisch medium met uniforme eigenschappen.
Intelligent ontwerpen
De vooruitgang die de scheikunde de laatste dertig jaar heeft geboekt (en nog steeds boekt) is voor een belangrijk deel te danken aan de mogelijkheden van de computermodellering. Het helpt chemici op een intelligente manier moleculen te ontwerpen – in plaats van maar wat te synthetiseren en ‘proberen of het werkt’.
Denk bijvoorbeeld aan zonnecellen uit kunststof, waarin speciale moleculen het licht ‘oogsten’. Met de computer zijn zulke moleculen op efficiëntie te screenen. Daarna zijn in het laboratorium de meest kansrijke exemplaren heel gericht te bestuderen. Een ander voorbeeld is de ontwikkeling van moleculen die als geneesmiddel in de stofwisseling van onze cellen moeten ingrijpen. Ook hier versnelt de computer de identificatie van moleculen met de juiste functionaliteit. Een ander voorbeeld is het ontwerp van katalysatoren voor efficiënte chemische processen – de lijst van toepassingen is haast eindeloos.

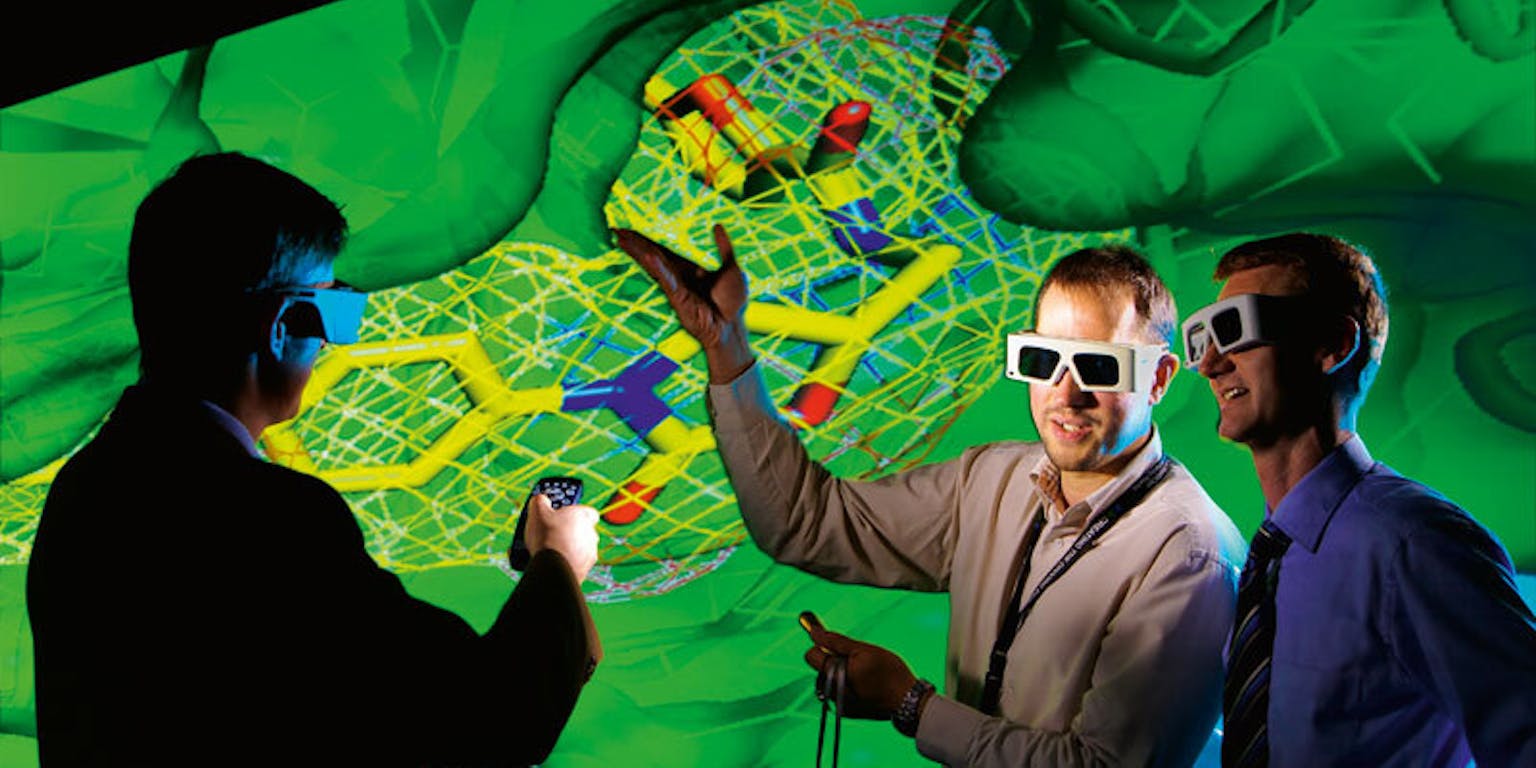
Met behulp van geavanceerde visualisatietechnieken stappen chemici als het ware in hun modellen. Hier werken onderzoekers van Bayer met een 3D-weergave van het actieve centrum van een eiwit.
Bayer PharmaOndertussen gaan in de computational chemie de ontwikkelingen ook steeds verder. Steeds slimmere modellen, almaar krachtiger computers: het eind is nog niet in zicht. De droom van Michael Levitt is een computer waarmee het mogelijk is het functioneren van complete levende organismen te doorgronden, gebaseerd op simulaties op moleculair niveau. Een gedurfde gedachte.